![]() The UK’s ambitious plan to sequence 100,000 whole genomes of NHS patients over the next 3-5 years, announced by the UK Prime Minister in December last year, sparked interest and curiosity throughout the UK genetics community. Undeterred by the enormity of the task, a new company, Genomics England Limited (GeL), was set up in June of this year by the Department of Health, tasked with delivering the UK100K genome project. Yesterday, they held what I’m sure will be the first of many ‘Town Hall’ engagement events, to inform and consult clinicians, scientists, patients and the public on their nascent plans.
The UK’s ambitious plan to sequence 100,000 whole genomes of NHS patients over the next 3-5 years, announced by the UK Prime Minister in December last year, sparked interest and curiosity throughout the UK genetics community. Undeterred by the enormity of the task, a new company, Genomics England Limited (GeL), was set up in June of this year by the Department of Health, tasked with delivering the UK100K genome project. Yesterday, they held what I’m sure will be the first of many ‘Town Hall’ engagement events, to inform and consult clinicians, scientists, patients and the public on their nascent plans.
So what did we learn? First, let’s be clear on the aims. GeL’s remit is to deliver 100,000 whole genome sequences of NHS patients by the end of 2017. No fewer patients, no less sequence. At its peak, GeL will produce 30,000 whole genome sequences per year. There’s no getting away from the fact that this is an extremely ambitious plan! But fortunately, the key people at GeL are under no illusions about the fact that theirs is a near impossible task. Continue reading ‘Genomics England and the 100,000 genomes’

 This week sees the publication of a large study of the genetics of multiple sclerosis. A consortium of 23 research groups gathered together data on nearly 10,000 MS suffers, and discovered 29 new genetic variants that contribute to disease risk. Overall, genetic variants for MS can now explain around 20% of the overall heritability of the disease, and these genetic variants highlight pathways that are likely to be important in the disease (such as T-helper-cell differentiation). Notably, this study is published in Nature, which is pretty rare for genome-wide association studies such as this. Perhaps related to this is the wonderful degree of detail included in the figures, such as in the ancestry plots of individuals in the study (see left). It is also surprisingly readable, containing just 4 pages of main article, with the nitty-gritty relegated to 100 pages of supplemental text. [LJ]
This week sees the publication of a large study of the genetics of multiple sclerosis. A consortium of 23 research groups gathered together data on nearly 10,000 MS suffers, and discovered 29 new genetic variants that contribute to disease risk. Overall, genetic variants for MS can now explain around 20% of the overall heritability of the disease, and these genetic variants highlight pathways that are likely to be important in the disease (such as T-helper-cell differentiation). Notably, this study is published in Nature, which is pretty rare for genome-wide association studies such as this. Perhaps related to this is the wonderful degree of detail included in the figures, such as in the ancestry plots of individuals in the study (see left). It is also surprisingly readable, containing just 4 pages of main article, with the nitty-gritty relegated to 100 pages of supplemental text. [LJ]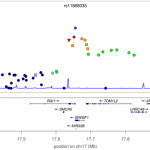



 Genomes Unzipped content is available for reuse under a Creative Commons Attribution-ShareAlike 3.0 Unported License.
Genomes Unzipped content is available for reuse under a Creative Commons Attribution-ShareAlike 3.0 Unported License. RSS
RSS Twitter
Twitter
Recent Comments