 This is a guest post by Peter Cheng and Eliana Hechter from the University of California, Berkeley.
This is a guest post by Peter Cheng and Eliana Hechter from the University of California, Berkeley.
Suppose that you’ve had your DNA genotyped by 23andMe or some other DTC genetic testing company. Then an article shows up in your morning newspaper or journal (like this one) and suddenly there’s an additional variant you want to know about. You check your raw genotypes file to see if the variant is present on the chip, but it isn’t! So what next? [Note: the most recent 23andMe chip does include this variant, although older versions of their chip do not.]
Genotype imputation is a process used for predicting, or “imputing”, genotypes that are not assayed by a genotyping chip. The process compares the genotyped data from a chip (e.g. your 23andMe results) with a reference panel of genomes (supplied by big genome projects like the 1000 Genomes or HapMap projects) in order to make predictions about variants that aren’t on the chip. If you want a technical review of imputation (and the program IMPUTE in particular), we recommend Marchini & Howie’s 2010 Nature Reviews Genetics article. However, the following figure provides an intuitive understanding of the process.
Imputation requires two ingredients: first, observed genotype data, often from a genotyping chip (in our example, your 23andMe results), and second, a “reference panel” (in our example, from the 1000 Genomes Project). Because the reference panel was produced by genome sequencing it contains genotypes from a much larger number of SNPs than your data (and notably, this denser SNP set includes the SNP of interest). If we look at a particular chunk of your genome (or “chromosome”), your data will look most similar to a chromosome from a person in the reference panel (in the figure, the chromosome coloured green). This is because at this part of the genome, you share a degree of (probably distant) common ancestry with this person. The imputation algorithm then fills in your genotypes at SNPs that weren’t on the chip using genotypes from this related reference individual.
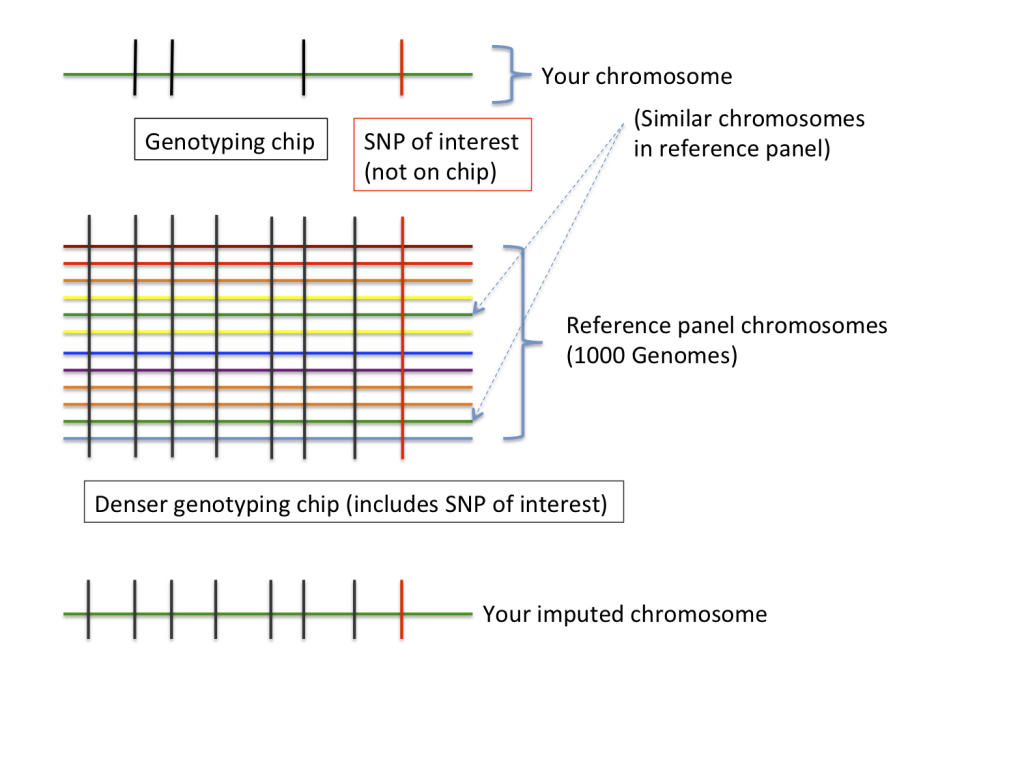
Different parts of your genome will be more closely related to different individuals in the reference set, so the imputation algorithm models your genome as a mosaic of related genomes, and uses these related genomes to fill in all of your missing data. Different imputation programs do slightly different things, but most give very similar results. In the examples we looked at, we used the imputation program IMPUTE2.
What does imputation buy you?
Genotype imputation can be useful in various ways. Say you are interested in knowing your genotype at specific SNPs (the COMT warrior/worrier variant in the NYT article linked to above, for instance). These SNPs of interest could be SNPs that are known to be linked with a certain trait or disease. If 23andMe doesn’t genotype the SNPs of interest, imputation can be used to obtain predictions for these variants. In addition, 23andMe periodically updates their genotyping chip and offers the consumer the chance to upgrade their data for a price. Instead of having to pay them to get your results updated, imputation allows you to predictively update your 23andMe results for free.
How accurate is IMPUTE2, that is, how often does it guess your genotype correctly? IMPUTE2 gives a sense of the accuracy of its “guesses”; for many SNPs, this confidence is >95% (more common SNPs tend to be imputed more accurately). To give a rough idea, the average confidence was 97.58% across all SNPs Peter imputed in his own genome. Although the average accuracy is impressive, there’s always a chance that wrong predictions will occur, or that the inaccuracies will coincide with SNPs of interest: around one in 40 SNP predictions will be wrong, and this is likely to be higher for SNPs that are less common in the population. Finally and importantly, imputation accuracy depends on your own ancestral population and how well it is represented in the reference panel (in the figure, this corresponds to how many green chromosomes exist in the 1000 Genomes data). The more closely related your “closest relatives” in the reference panel are, the more accurate the imputation results will be.
If you decide to use IMPUTE2 to update your 23andMe results, remember that this “update” is only as good as the imputation accuracy, while 23andMe’s genotyping has the accuracy of a genotype chip (i.e. very high). There is a trade-off in imputation: it’s free and easy to do, but we are not guaranteed each prediction is correct, so results should be taken with a grain of salt.
Note from Eliana: In Fall Semester of 2011, I taught Math 127 at Berkeley as a class on computational methods in personal genomics. All of the students in the course had the opportunity to have their genomes sequenced by 23andMe, and their final projects centered on ways to extend the 23andMe analysis to their own data. Peter was an undergraduate student in the class. Peter’s final project involved writing a script that transforms the 23andMe genotypes.txt raw data file to a format that is compatible with the imputation program IMPUTE2 (the script is available here). I used Peter’s program to impute my BRCA2 (breast cancer susceptibility) variants not included on the 23andMe chip, and thought others might be interested in similar analyses. (There is a whole separate discussion about what imputation means for gene patents, but we’re going to leave that be for this post.)








 RSS
RSS Twitter
Twitter
great post! definitely going to try and play around with imputation this weekend.
For those interested in imputing their data without using command line scripts, Interpretome (http://www.interpretome.com) has a simple imputation function. When you look up variants that are not on your chip, they are automatically imputed (using Hapmap data) where possible. No high-throughput imputation (for that you’ll need to use a script like one of these).
konrad, who doesn’t have an interest in using command line scripts? :-) though i’ll be promoting that feature of interpretome perhaps this wkd. i’m interested in function all of a sudden after my wife’s discovery about my daughter yesterday….
Imputation can be dangerous. It is well known the calls for the APOE variants are broken in the HapMap project (1000g project is fine though).
Thanks for the script. I just downloaded my raw files and I tried running the script, but I receive an error:
Search pattern not terminated at format_for_impute.pl line 221
anyone knows the solution?
Thanks!
Erik
If you want a quick and simple way to impute single variants starting from 23andMe raw data (as for example the rs429358 APOE SNP, not genotyped in the 23andMe v2 chip), I came up with a couple of scripts that will do this in no time (that is, less than a minute) using Beagle 4 and the 1000 Genomes Project reference panel. The instructions (which require running a couple of command line scripts) can be found at http://apol1.blogspot.com/2013/08/impute-apoe-and-apol1-with-23andme.html and do not require you to upload your genotype data to any server.