Last week, scientists at the European Molecular Biology Laboratory reported that they had sequenced the genome of the Henrietta Lacks, or “HeLa”, cell line. This report was met with considerable consternation by those who (justifiably, in my opinion) wondered why scientists are still experimenting on a cell line obtained without consent in the 1950s [1]. In response to a bit of a backlash, the researchers removed the HeLa sequence from the public internet, and even the paper itself might disappear from the formal scientific literature.
However, it is unfair to treat the authors of this paper as scapegoats for the systematic failure of scientists to deal with issues surrounding genomic “privacy”. Consider this important piece of information: the genome sequence of the HeLa cell line has been publicly available for years (and remains so).
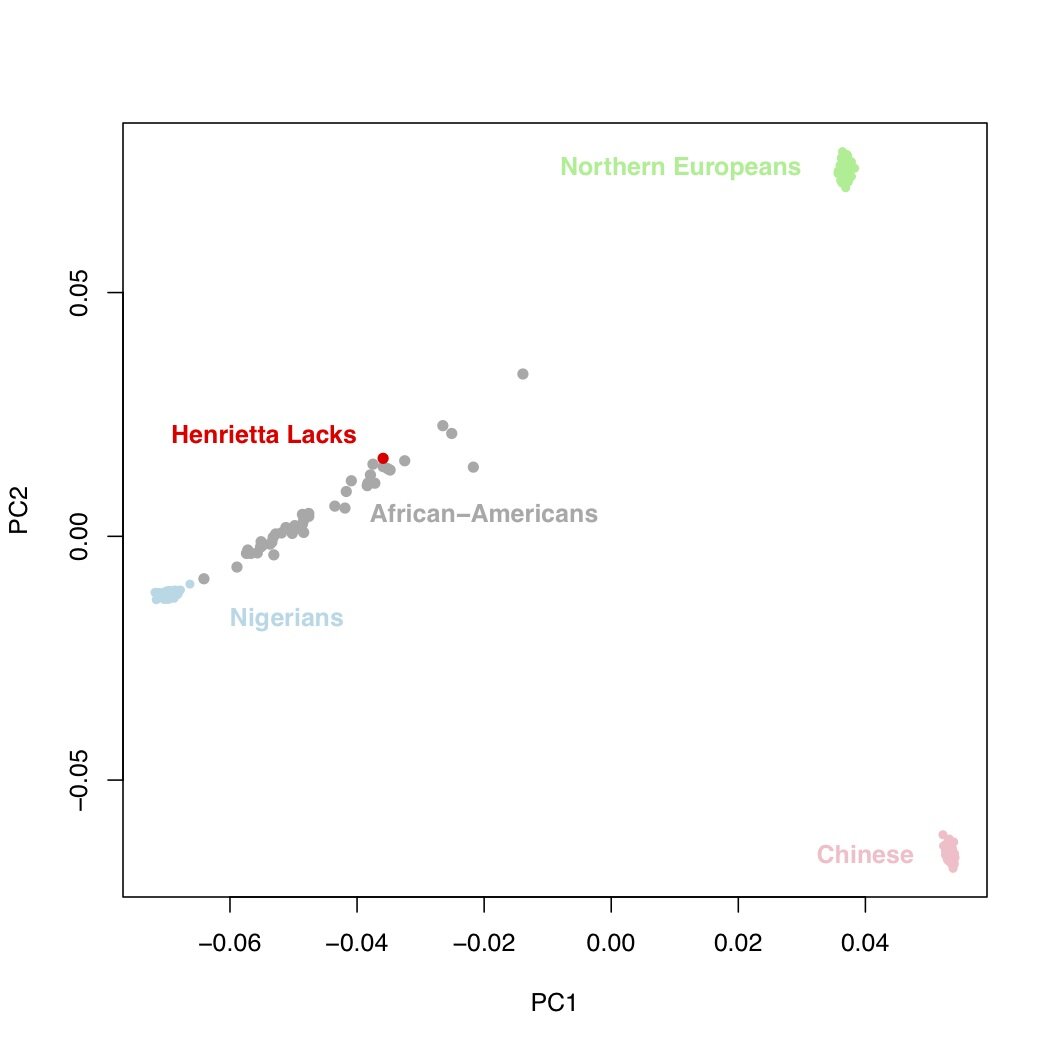
Continue reading ‘Henrietta Lacks’s genome sequence has been publicly available for years’
 This is a guest post by Peter Cheng and Eliana Hechter from the University of California, Berkeley.
This is a guest post by Peter Cheng and Eliana Hechter from the University of California, Berkeley.


 RSS
RSS Twitter
Twitter
Recent Comments